Computerchemie: hoe computers moleculen voorspellen en medicijnen ontwerpen
Ontdek hoe computerchemie molecuulstructuren voorspelt en innovatieve geneesmiddelen ontwerpt — van simulaties tot AI-gestuurde ontwikkeling.
De computerchemie is een tak van de chemie die gebruikmaakt van technieken uit de de computerwetenschap om chemische problemen te analyseren en op te lossen. Met speciale software berekent men de structuren en eigenschappen van moleculen en vaste stoffen. Computerchemie vult de informatie aan die uit laboratorium-experimenten komt en kan chemische fenomenen voorspellen die nog niet direct zijn waargenomen. Daardoor is het een belangrijk instrument bij het ontwerp van nieuwe geneesmiddelen en materialen.
Afbeeldingengalerij
8 Afbeeldingen


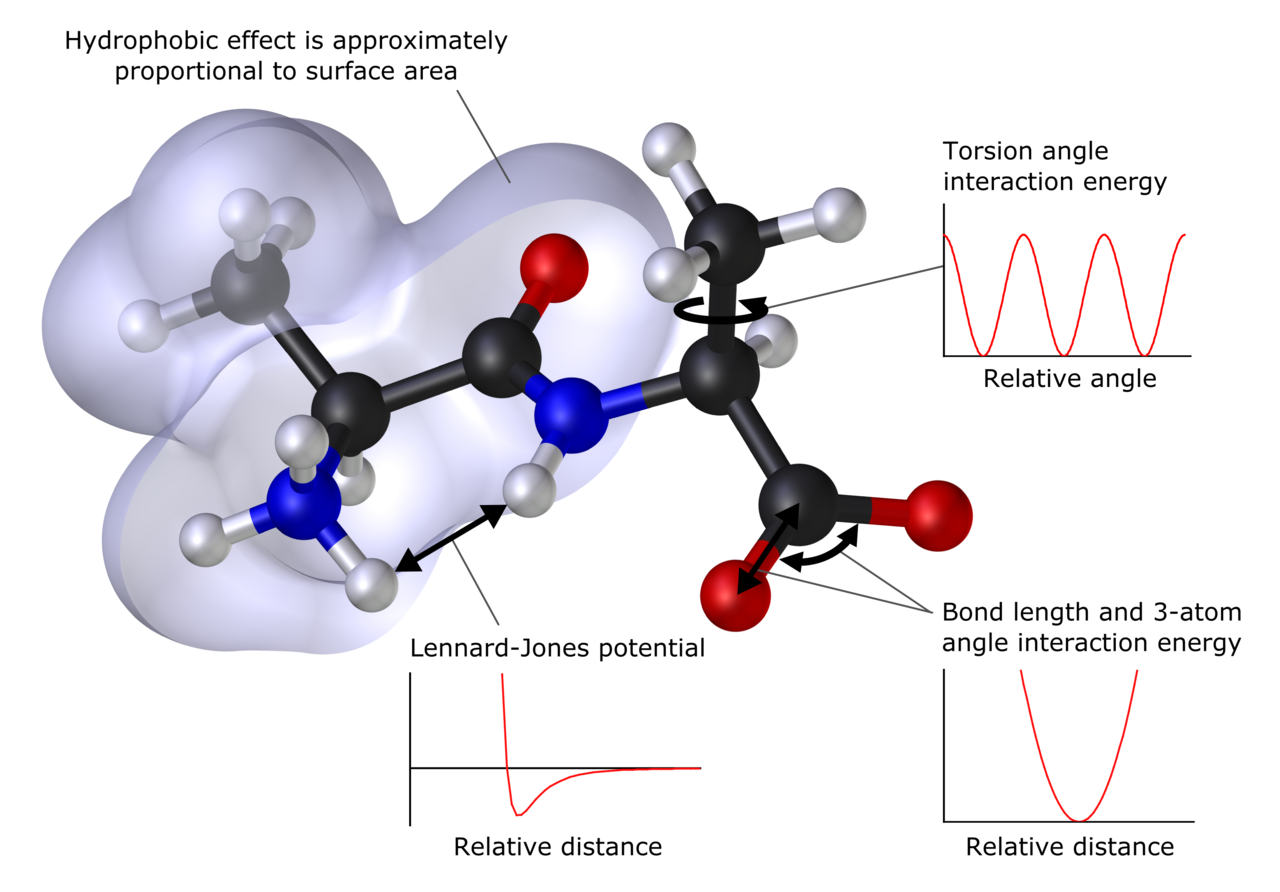


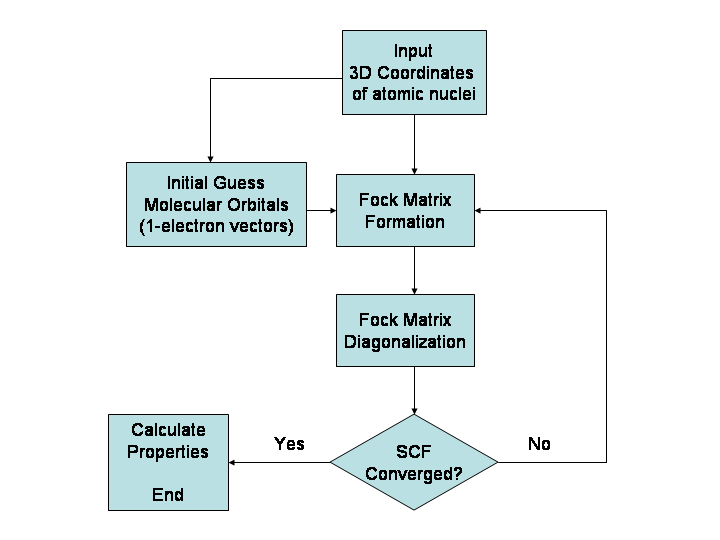
Wat kan computerchemie voorspellen?
Computerchemische methoden geven inzicht in veel verschillende grootheden. Ze voorspellen onder meer de verwachte posities van de atomen van een molecuul (de structuur), absolute en relatieve (interactie‑) energieën, de elektronische ladingsverdeling, dipolen en hogere meerpolige momenten, trillingsfrequenties, reactiviteit en diverse spectroscopische grootheden. Verder kunnen sommige methoden ook doorsneden voor botsingen met andere deeltjes en dynamische eigenschappen voorspellen, zoals transport en diffusie.
Belangrijke methoden en technieken
- Quantumchemie (ab initio): rekent op de Schrödingervergelijking en omvat technieken als Hartree–Fock, MP2 en coupled cluster (CC). Deze methoden zijn vaak zeer nauwkeurig maar ook rekenslimitief.
- Density Functional Theory (DFT): een efficiënte benadering voor de elektronische structuur, veel gebruikt voor moleculen en vaste stoffen; levert een goede balans tussen nauwkeurigheid en computerkosten.
- Semi‑empirische methoden: versimpelde quantummethoden die parameters uit experiment of hogere berekeningen gebruiken; snel maar minder algemeen nauwkeurig.
- Moleculaire mechanica (force fields): behandelt atomen als deeltjes verbonden door veren en gebruikt empirische krachtvelden (bijv. AMBER, CHARMM, OPLS) — geschikt voor grote biologische systemen en langdurige simulaties.
- Moleculaire dynamica (MD) en Monte Carlo (MC): simuleren de beweging van atomen en moleculen in de tijd of monsteren toestanden, cruciaal voor het bestuderen van conformatieveranderingen, bindingsdynamica en thermodynamica.
- Binding- en farmaceutisch ontwerp: docking, virtuele screening, QSAR en vrije-energie‑berekeningen (FEP, MM‑PBSA) helpen bij het selecteren en optimaliseren van kandidaat‑geneesmiddelen.
Toepassingen in geneesmiddelenontwerp en materialen
In het ontwerp van geneesmiddelen wordt computerchemie gebruikt om mogelijke bindingsplaatsen, bindingssterkte en selectiviteit in eiwitten te voorspellen. Workflowstappen zijn onder meer:
- virtueel screenen van grote moleculenbibliotheken;
- moleculaire docking om mogelijke bindingsorientaties te vinden;
- geavanceerde vrije-energieberekeningen voor nauwkeurige affiniteitsschatting;
- optimalisatie van ADMET‑eigenschappen met behulp van voorspellende modellen.
Voor materialen helpt computerchemie bij het voorspellen van kristalstructuren, elektronische eigenschappen, stabiliteit en transporteigenschappen, en bij het ontwikkelen van materialen met gewenste mechanische, optische of elektronische kenmerken.
Schaalbaarheid, kosten en beperkingen
De rekenbelasting neemt sterk toe met de grootte van het systeem. Nauwkeurige quantummethoden schalen vaak slecht (soms als een hoge macht van het aantal atoomorbitalen), terwijl DFT en semi‑empirische methoden minder zwaar zijn. Moleculaire mechanica en deels machine learning‑methoden schalen veel beter en kunnen systemen met tienduizenden tot miljoenen atomen behandelen. Keuze van de methode is altijd een compromis tussen nauwkeurigheid en berekenbaarheid.
Software, hardware en praktische aspecten
Voor computerchemie bestaan veel specialistische pakketten (zowel open source als commercieel). Rekentaken profiteren van moderne hardware: multi‑core CPU's, GPU's, clusters en cloud‑resources versnellen berekeningen aanzienlijk. Belangrijke praktische punten zijn modelvalidatie aan experimenten, juiste keuze van parameters/force fields, en reproducibiliteit van berekeningen.
Nieuwe ontwikkelingen: AI en machine learning
Machine learning versnelt en verbetert veel computerchemische toepassingen. Voorbeelden:
- snelle voorspellers voor energieën en eigenschappen die duurdere DFT‑berekeningen vervangen;
- generatieve modellen die nieuwe moleculen ontwerpen met gewenste eigenschappen;
- surrogaatmodellen voor vrije‑energieberekeningen en optimalisatie van reacties.
AI-methoden worden vaak gecombineerd met traditionele berekeningen en experimenten om zoekruimten efficiënter te doorlopen.
Workflow, validatie en betrouwbaarheid
Een goede computerchemische studie bevat doorgaans: helder gedefinieerde vraagstelling, selectie van geschikte methoden, convergentiecontroles, vergelijking met beschikbare experimentele data en tenslotte onzekerheidsanalyse. Validatie aan experimenten blijft cruciaal om voorspellingen betrouwbaar te maken.
Samengevat: computerchemie is een veelzijdig vakgebied dat van grote waarde is voor het begrijpen en voorspellen van moleculaire eigenschappen en reacties. Het verbindt theorie en experiment en speelt een centrale rol bij moderne ontwikkelingen in geneesmiddelen en materialen, met voortdurende vooruitgang dankzij verbeterde algoritmes en kunstmatige intelligentie.

Gerelateerde pagina's
- Bio-informatica
- Statistische mechanica
Vragen en antwoorden
V: Wat is computationele chemie?
A: Computationele chemie is een tak van de chemie die computerwetenschappen gebruikt om chemische problemen op te lossen. Het kan worden gebruikt om de structuren en eigenschappen van moleculen en vaste stoffen te berekenen, chemische verschijnselen te voorspellen die nog niet zijn waargenomen, en nieuwe geneesmiddelen en materialen te ontwerpen.
V: Naar welke soorten systemen kijkt de computationele chemie?
A: Computationele chemie bekijkt zowel statische als dynamische systemen. Het systeem kan bestaan uit één molecuul, een groep moleculen of een vaste stof.
V: Welke soorten informatie kan computationele chemie verschaffen?
A: Computationele chemie kan informatie verschaffen zoals structuur (posities van atomen), absolute en relatieve energieën, elektronische ladingsverdelingen, dipolen en hogere multipoolmomenten, trillingsfrequenties, reactiviteit of andere spectroscopische grootheden, en doorsneden voor botsingen met andere deeltjes.
V: Hoe nauwkeurig zijn de methoden die in de computationele chemie worden gebruikt?
A: De nauwkeurigheid van de in de computationele chemie gebruikte methoden varieert van zeer nauwkeurig tot zeer approximatief. Zeer nauwkeurige methoden zijn doorgaans alleen haalbaar voor kleine systemen.
V: Hoe vult computationele chemie experimentele gegevens aan?
A: Computationele chemie is normaal gesproken een aanvulling op de informatie die door chemische experimenten wordt verkregen. Zij kan worden gebruikt om resultaten te voorspellen die nog niet experimenteel zijn waargenomen.
V: Heeft de grootte van het te bestuderen systeem invloed op de benodigde computertijd?
A: Ja - naarmate de omvang van het bestudeerde systeem toeneemt, neemt ook de hoeveelheid computertijd die nodig is voor de analyse toe, evenals de middelen zoals geheugen en schijfruimte die nodig zijn voor de opslag.
Gerelateerde artikelen
Auteur
AlegsaOnline.com Computerchemie: hoe computers moleculen voorspellen en medicijnen ontwerpen Leandro Alegsa
URL: https://nl.alegsaonline.com/art/22297