Amyotrofische laterale sclerose (ALS): definitie, oorzaken en symptomen
Leer wat ALS is: oorzaken, symptomen en verloop van amyotrofische laterale sclerose, herken vroege signalen, diagnostiek en impact op patiënten en naasten.
Amyotrofische laterale sclerose (ALS) is een vorm van motorische neuronziekte en wordt in het Engels ook vaak de ziekte van Lou Gehrig genoemd. Het is een chronische, progressieve en meestal dodelijke neurologische aandoening waarbij vooral de motorische zenuwcellen (neuronen) in het centrale zenuwstelsel langzaam afsterven. Daardoor verliezen mensen geleidelijk het vermogen om vrijwillige spieren aan te sturen: lopen, spreken, slikken en ademhalen worden steeds moeilijker.
Motorische neuronen zijn de zenuwcellen die boodschappen van de hersenen en het ruggenmerg naar de spieren sturen. Bij ALS sterven zowel de bovenste motorische neuronen (in de hersenen) als de onderste motorische neuronen (in het ruggenmerg en de hersenstam) af. Dit leidt tot spierzwakte, krampen, spierstijfheid en spierverlies (atrofie). De aandoening verergert meestal in de loop van maanden tot enkele jaren, en veroorzaakt toenemende invaliditeit en uiteindelijk de dood, vaak door ademhalingsfalen. Niet-bewegende functies zoals hartslag en spijsvertering blijven meestal intact omdat die autonoom worden gereguleerd.
ALS is de meest voorkomende vorm binnen de groep van motorische neuronziekten. Bij ongeveer 5–10% van de patiënten is er sprake van een erfelijke vorm; deze gevallen zijn geërfd van één of beide ouders. De overige 90–95% zijn sporadisch, zonder een duidelijk erfelijke oorzaak.
Afbeeldingengalerij
10 Afbeeldingen





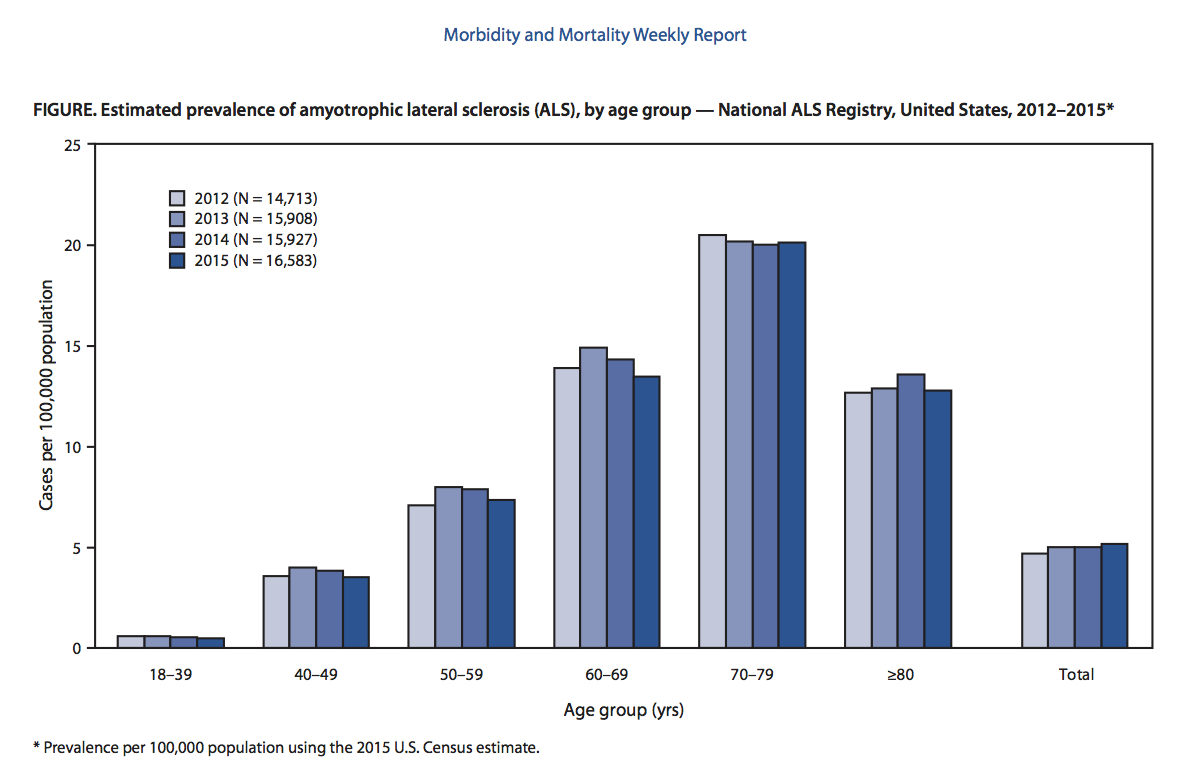



Oorzaken en risicofactoren
De precieze oorzaak van ALS is niet volledig bekend. Waarschijnlijk spelen meerdere factoren een rol, waaronder genetische veranderingen, ontregelde eiwitafbraak in cellen, oxidatieve stress en ontstekingsprocessen. Bekende genetische oorzaken omvatten mutaties in genen zoals SOD1, C9orf72 en anderen (genetische diagnostiek kan bij familiegeschiedenis worden overwogen).
Bekende risicofactoren zijn onder meer:
- Leeftijd (meestal begin tussen 50–70 jaar)
- Man zijn (licht verhoogd risico)
- Familiaire voorgeschiedenis van ALS of soortgelijke ziekten
- Externe factoren zoals zware fysieke belasting, bepaalde toxines of spierstress worden onderzocht maar geven geen duidelijke oorzaak
Veelvoorkomende symptomen
ALS kan op verschillende manieren beginnen (bijvoorbeeld met moeite bij lopen of met spreken). Symptomen treden doorgaans gradueel op en verslechteren met de tijd. Veel voorkomende klachten zijn:
- Spierzwakte in armen of benen (valt vaker eerst één ledemaat op)
- Slik- of spreekproblemen (dysfagie, dysartrie)
- Spierkrampen en fasciculaties (spiertrekkingen)
- Stijfheid en verhoogde spierreflexen (spasticiteit)
- Afname van spiermassa (atrofie)
- Ademhalingsproblemen bij ziektevorderingen
Sensorische functies zoals gevoel, en vaak ook de meeste autonome functies, blijven relatief gespaard. Veel patiënten behouden hun intelligentie en geheugen, maar bij een deel van de patiënten treden cognitieve en gedragsveranderingen op en soms ontwikkelt zich een frontotemporale dementie met veranderingen in persoonlijkheid en gedrag.
Diagnose
Er is geen enkele test die ALS definitief bewijst; de diagnose wordt gesteld op basis van het verhaal van de patiënt, lichamelijk en neurologisch onderzoek en aanvullende onderzoeken om andere oorzaken uit te sluiten. Gebruikelijke onderzoeken zijn:
- Electromyografie (EMG) en zenuwgeleidingsonderzoeken om de functie van motorische zenuwen en spieren te beoordelen
- Magnetische resonantiebeeldvorming (MRI) van hoofd en ruggenmerg om andere aandoeningen uit te sluiten
- Bloedonderzoek en soms liquoronderzoek om metabole, infectieuze of inflammatoire oorzaken te onderzoeken
- Genetisch onderzoek bij vermoeden van erfelijke vorm of familiegeschiedenis
Behandeling en zorg
Er bestaat momenteel geen genezing voor ALS, maar er zijn behandelingen die het ziekteverloop licht kunnen vertragen en vooral gericht zijn op het verbeteren van kwaliteit van leven en het behandelen van symptomen. Belangrijke onderdelen van zorg:
- Medicatie: riluzol is aangetoond de overleving iets te verlengen; in sommige landen is ook edaravone beschikbaar voor een subset van patiënten
- Multidisciplinaire zorg: samenwerking tussen neurolog, longarts, revalidatiearts, diëtist, logopedist, fysiotherapeut en ergotherapeut verbetert functie en kwaliteit van leven
- Logopedie en sliktraining; bij ernstige slikproblemen kan een voedingssonde (gastrostomie) nodig zijn
- Ademhalingsondersteuning: niet-invasieve ventilatie (NIV) kan de levenskwaliteit en de overleving verbeteren
- Spier- en pijnbestrijding, spasticiteitsbeheer (medicatie, fysiotherapie)
- Palliatieve zorg en begeleiding voor patiënt en familie, inclusief psychosociale en praktische steun
Prognose
Het beloop van ALS is zeer variabel. Gemiddeld ligt de overleving na diagnose rond 3–5 jaar, maar sommige mensen leven veel langer, anderen korter. Begin in de hersenstam (bulbaire vorm) of snelle ademhalingsinsufficiëntie gaan vaak samen met een sneller beloop. Vroege multidisciplinaire zorg en ademhalingsondersteuning kunnen de levenskwaliteit en soms de duur verbeteren.
Wanneer naar de arts
Zoek medische aandacht bij aanhoudende of toenemende spierzwakte, moeite met spreken, slikken of ademen. Vroege evaluatie door een neuroloog is belangrijk om andere behandelbare oorzaken uit te sluiten en tijdig multidisciplinaire zorg te starten.
Samenvatting
ALS is een ernstige, progressieve motorische neuronziekte met verlies van vrijwillige spierfunctie door degeneratie van bovenste en onderste motorische neuronen. Er is geen genezing, maar vroegtijdige diagnose en een geïntegreerde behandel- en zorgaanpak kunnen complicaties verminderen en de kwaliteit van leven verbeteren. Bij familiegeschiedenis kan erfelijkheid een rol spelen; overleg met uw arts over genetisch onderzoek indien relevant.
Symptomen
Motorische neuronziekte vertoont niet veel symptomen, waardoor het erg moeilijk is om een diagnose te stellen. Het treft meestal mensen in de leeftijd van 40-60 jaar. De vroegste symptomen kunnen zijn: stuiptrekkingen, krampen of stijfheid van de spieren, spierzwakte van een arm of een been, een vertroebelde en vreemd klinkende neusspraak, of een moeilijke tijd van kauwen of slikken. Deze algemene symptomen ontwikkelen zich dan tot duidelijke zwakte of atrofie die een arts kan doen geloven dat een persoon ALS heeft.
De delen van het lichaam die getroffen worden door de vroege symptomen van ALS zijn afhankelijk van welke spieren in het lichaam het eerst worden aangetast. Ongeveer 75% van de mensen heeft een beginnende ALS. In sommige van deze gevallen hebben de symptomen eerst invloed op één van de benen en hebben patiënten last van onhandigheid bij het lopen of rennen of merken ze dat ze vaker struikelen of struikelen. Andere patiënten die aan een ledemaat beginnen, zien eerst de effecten van de ziekte op een hand of arm, omdat ze een moeilijke tijd hebben met eenvoudige taken die handvaardigheid vereisen, of het vermogen om kleine dingen te bewegen, zoals het dichtknopen van een hemd, het schrijven of het draaien van een sleutel in een slot.
Ongeveer 25% van de gevallen zijn bulbaire beginnende ALS. Deze patiënten hebben eerst moeite om duidelijk te spreken. Spraak wordt moeilijk te begrijpen en vertroebeld. Spreken door de neus en zachter praten zijn vaak de eerste symptomen. Moeite met slikken en verlies van de tongbeweging volgen. Uiteindelijk is er sprake van totaal verlies van spraak en het vermogen om de luchtweg vrij te houden bij het slikken.
Behandeling
Er is geen remedie voor ALS. De Food and Drug Administration (FDA) heeft echter de eerste medicijnbehandeling voor de ziekte goedgekeurd: riluzole. Riluzole wordt verondersteld de schade aan de motorische neuronen te verminderen. Klinische studies met ALS-patiënten toonden aan dat riluzol een betere overlevingskans van enkele maanden geeft, vooral bij mensen die moeite hebben met slikken. Het medicijn geeft ook meer tijd voordat een patiënt hulp nodig heeft bij de ademhaling door middel van een beademingsapparaat of tracheotomie. Riluzol geneest niet de schade die al is aangericht aan de motorische neuronen.
Andere behandelingen voor ALS zijn bedoeld om de symptomen minder pijnlijk te maken en de levenskwaliteit van de patiënten te verbeteren.
Vragen en antwoorden
V: Wat is de neuronziekte?
A: De ziekte van Motor neuronen is een chronische, progressieve en dodelijke neurologische ziekte die het afsterven van zenuwcellen in het centrale zenuwstelsel veroorzaakt, wat leidt tot toenemende invaliditeit en uiteindelijk de dood.
V: Wat doen de zenuwcellen die aangetast zijn door de neuronziekte?
A: De zenuwcellen die aangetast zijn door de motorneuronziekte controleren vrijwillige spierbewegingen, zoals spreken, lopen, slikken en het bewegen van het lichaam.
V: Bestaat er een behandeling voor de neuronziekte?
A: Nee, er is geen genezing bekend voor de neuronziekte.
V: Wat is amyotrofische laterale sclerose (ALS)?
A: Amyotrofische laterale sclerose (ALS) is de meest voorkomende vorm van de neuronziekte die spierzwakte en krimpende spieren in het hele lichaam veroorzaakt, wat leidt tot spieratrofie.
V: Wat veroorzaakt de spieratrofie bij ALS?
A: Zowel de bovenste motorneuronen als de onderste motorneuronen sterven af, waardoor ze geen berichten meer naar de spieren sturen, wat spieratrofie tot gevolg heeft.
V: Kan de ziekte van motorische neuronen erfelijk zijn?
A: Ja, ongeveer 5 tot 10% van de gevallen van motorneuronziekte wordt rechtstreeks geërfd van iemands ouders.
V: Heeft de neuronziekte invloed op iemands intelligentie, geheugen en persoonlijkheid?
A: Zelfs patiënten in latere stadia van de motorneuronziekte kunnen nog steeds dezelfde intelligentie, hetzelfde geheugen en dezelfde persoonlijkheid hebben als voordat de ziekte begon.
Gerelateerde artikelen
Auteur
AlegsaOnline.com Amyotrofische laterale sclerose (ALS): definitie, oorzaken en symptomen Leandro Alegsa
URL: https://nl.alegsaonline.com/art/66919
Bronnen
- ninds.nih.gov : "Motor neuron diseases fact sheet: National Institute of Neurological Disorders and Stroke (NINDS)"