Sikkelcelziekte
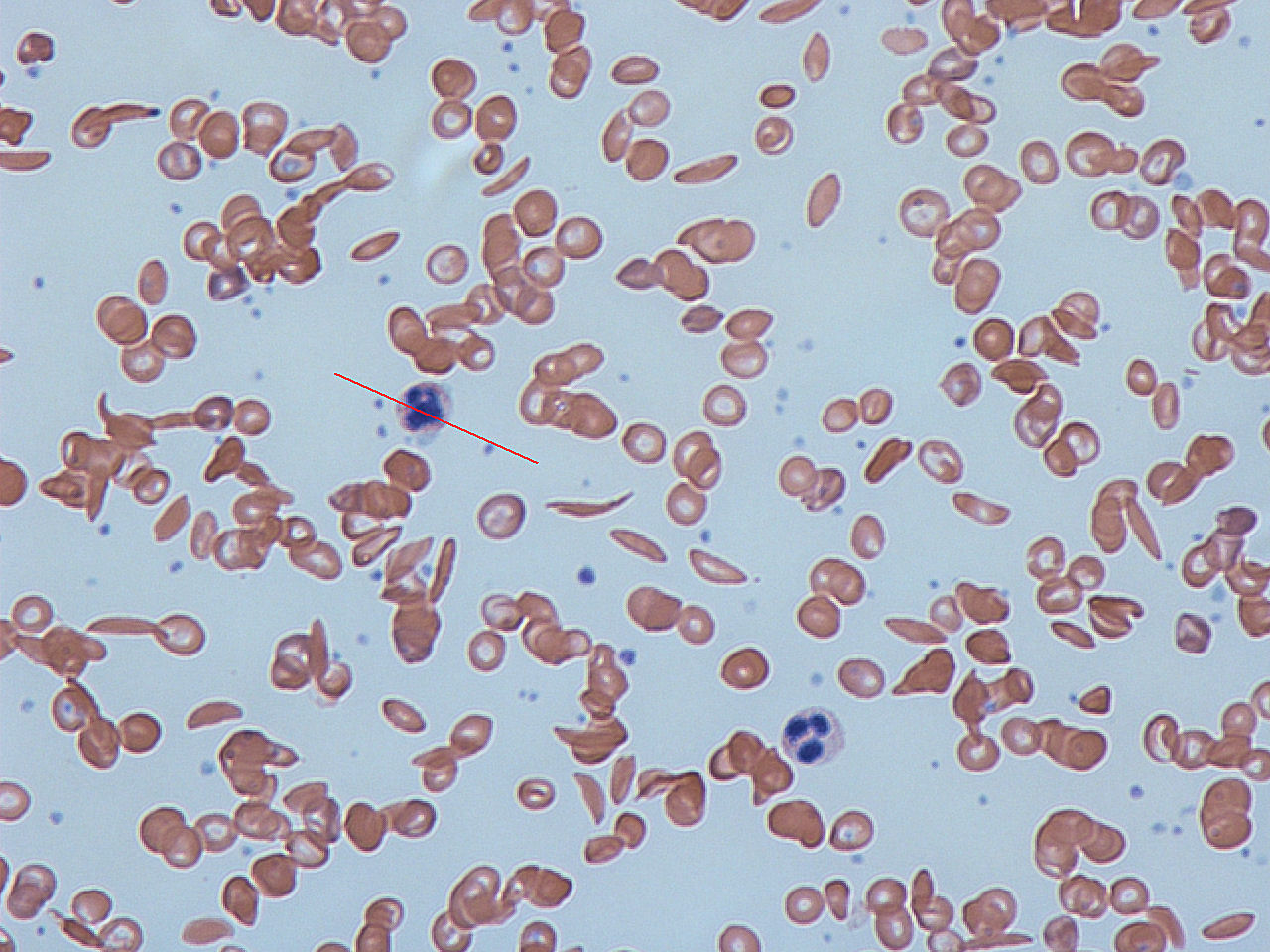
Sikkelcelanemie is een genetische ziekte. Het tast de rode bloedcellen aan. Het verandert de cellen van flexibele schijven in stijve halvemaantjes. Als veel rode cellen deze vorm aannemen raken aderen verstopt. Dit kan schade veroorzaken aan vel…
Sikkelcelanemie is een genetische ziekte. Het tast de rode bloedcellen aan. Het verandert de cellen van flexibele schijven in stijve halvemaantjes. Als veel rode cellen deze vorm aannemen raken aderen verstopt. Dit kan schade veroorzaken aan vele organen. De schade aan organen neemt toe met de tijd en leidt tot een vroege dood.
Afbeeldingengalerij
8 Afbeeldingen






De ziekte
Dit is een levenslange ziekte die in de kindertijd begint. De rode bloedcellen nemen een abnormale, stijve, sikkelvormige vorm aan. De cellen worden ook plakkerig. Dit veroorzaakt een moeilijke bloedstroom wanneer de cellen door lange smalle haarvaten stromen. Zuurstofgebrek vergroot het probleem. Als ze door zuurstofarme gebieden gaan, nemen de meeste cellen deze vorm aan. De cellen blijven dan plakken aan de binnenwand van bloedvaten, vooral aan de vertakking van aderen. Dit leidt tot een blokkade van de bloedstroom in veel organen. Ernstige complicaties kunnen het gevolg zijn.
Het klassieke voorbeeld van een sikkelcelcrisis is het "acuut borstsyndroom" (ACS). Dit is uniek voor sikkelcelpatiënten en kan binnen een dag of twee de dood tot gevolg hebben, tenzij het wordt behandeld. Historisch werd acuut borstsyndroom beschouwd als verschillend van infectie (longontsteking). Maar bij de behandeling heeft het weinig zin dat onderscheid te maken.
ACS is een klinische diagnose, geholpen door ten minste een röntgenfoto van de borstkas. In alle andere organen veroorzaakt zuurstofgebrek verwijding van de bloedvaten. Maar de long is een uniek orgaan waar de bloedvaten nauwer worden als er weinig zuurstof is. Dit unieke probleem maakt de long tot een belangrijk doelwit van de ziekte. Koorts is het meest voorkomende symptoom van ACS bij kinderen omdat infectie vaker voorkomt. Bij volwassenen kunnen circulerende stolsels en kapotte stukjes beenmerg ook bijdragen tot de verstopping van de vaten in de long en leiden tot ACS. ACS kan gedeeltelijk worden behandeld met een bloedtransfusie om de verziekte cellen te verdunnen met wat normale rode bloedcellen. Een nog betere behandeling is een procedure die uitwisseling van rode bloedcellen wordt genoemd. Automatische aferesemachines kunnen RBC-uitwisseling uitvoeren.
Een milder en vaker voorkomend probleem is "pijnlijke crisis". Bij een pijnlijke crisis gaat het om pijn in de flank, de rug en de dijen die door behandeling kan worden verlicht. Een pijnlijke crisis kan evolueren naar ergere problemen zoals acuut falen van de borstkas en andere organen, bv. beroertes, hartaanvallen. Zowel beroertes als hartaanvallen zijn algemene problemen die bij oudere mensen kunnen voorkomen. Maar bij sikkelpatiënten kunnen ze zelfs bij jongeren voorkomen.
De milt is verschillend betrokken bij verschillende etnische groepen met deze ziekte. Milt is het orgaan dat oude RBC's filtert en vernietigt. Oude RBC's zijn stijf en kunnen niet door een aantal zeer nauwe spleten in de milt. Maar bij sikkelpatiënten worden alle cellen heel snel stijf en blijven zo de milt verstoppen. Vanaf zeer jonge leeftijd sterven delen van de milt af door dit probleem. In de zuivere vorm van deze ziekte is de hele milt dood en verschrompeld voordat de patiënt volwassen is. Een normale milt bewaart een grote voorraad B-cellen die antilichamen maken en ons beschermen tegen bacteriën. Verlies van een werkende milt leidt tot verlies van bescherming tegen dergelijke bacteriën.
Bij veel Aziatische bevolkingsgroepen komt bèta thalassemie samen met sikkelcelziekte voor. Thalassemie zelf is een andere vorm van bloedarmoede. Maar de aard van de twee ziekten zijn tegengesteld. Thalassemie verhoogt de flexibiliteit van de rode cellen. Thalassemie zelf kan echter een ernstige ziekte zijn.
Populatiegenetica
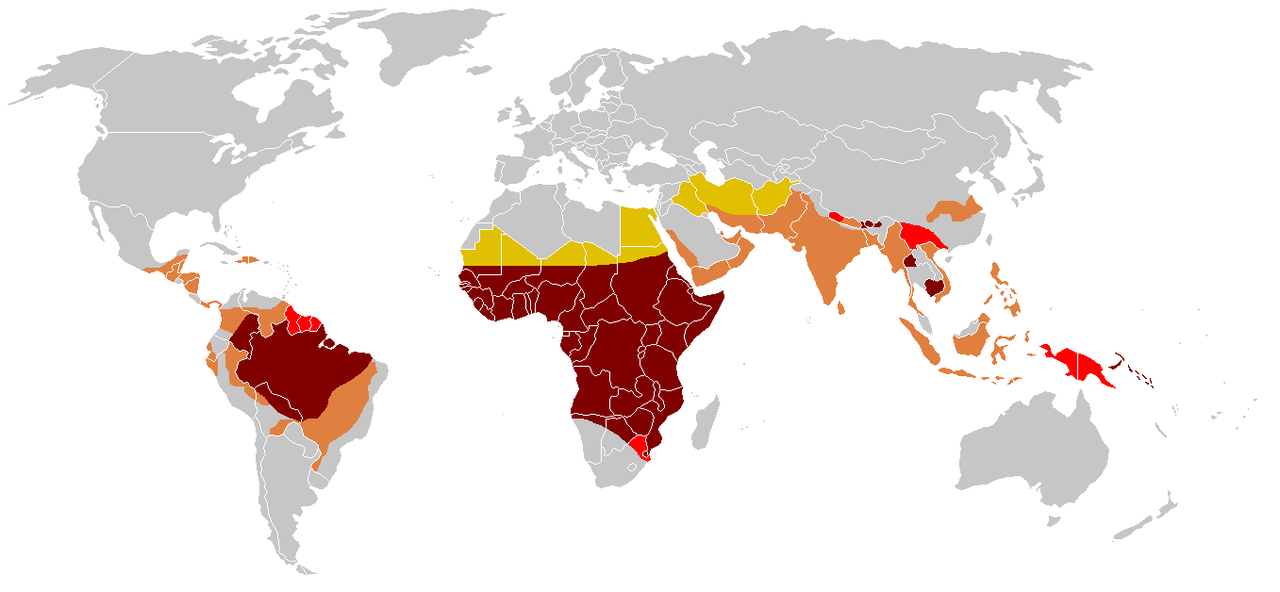
De sikkelziekte ontstaat door een enkele puntmutatie in het gen voor de bèta-keten van hemoglobine. Sikkelcelziekte komt vaker voor bij mensen (of hun nakomelingen) uit delen van tropische en subtropische gebieden waar malaria veel voorkomt of voorkwam. Een kwart van alle mensen van Afrikaanse afkomst ten zuiden van de Sahara draagt het gen.
We erven allemaal twee kopieën (allelen) van het hemoglobine bèta gen. Van elke ouder één. Sommige mensen zijn heterozygoot: zij hebben de sikkelmutatie in één exemplaar en het andere exemplaar is normaal. Zulke mensen worden sikkel trait of drager genoemd. Mensen met sikkelziekte zijn beter bestand tegen malaria dan normale mensen.
Wanneer beide allelen van een gen gelijk zijn (homozygoot) heeft een persoon gemuteerde exemplaren of beide normaal. Als beide allelen de mutatie hebben, veroorzaakt dat de volledige ziekte. In malariagevoelige gebieden sterven normale mensen vaak aan malaria, vaak voordat ze kinderen hebben gekregen. Degenen met beide kopieën met sikkelmutatie sterven aan sikkelziekte voordat zij zich kunnen voortplanten. Maar de heterozygoten hebben een betere overleving (en krijgen meer kinderen) dan beide homozygote groepen. De overerving van de ziekte is dus een voorbeeld van "heterozygoot voordeel".
Bij de volledige (homozygote) ziekte is de levensverwachting korter; in premoderne samenlevingen is de ziekte zelfs bijna fataal. Sir Cyril Clarke zei (verwijzend naar Oost-Afrika in de jaren 1960) "Bijna alle kinderen (met sikkelcelziekte) zullen op jonge leeftijd sterven".p25 . Studies in de moderne VS melden een gemiddelde levensverwachting van 42 jaar voor mannen en 48 jaar voor vrouwen.



Gerelateerde artikelen
Auteur
AlegsaOnline.com Sikkelcelziekte Leandro Alegsa
URL: https://nl.alegsaonline.com/art/90200
Bronnen
- nejm.org : nejm.org/doi/full/10.1056/NEJM200006223422502
- jci.org : "The impact of malaria parasitism: from corpuscles to communities"
- doi.org : 10.1172/JCI38307
- ncbi.nlm.nih.gov : 2735907
- pubmed.ncbi.nlm.nih.gov : 19729847
- content.nejm.org : "Mortality in sickle cell disease. Life expectancy and risk factors for early death"
- doi.org : 10.1056/NEJM199406093302303
- worldcat.org : 0028-4793
- pubmed.ncbi.nlm.nih.gov : 7993409