Pulmonale hypertensie (PAH): symptomen, oorzaken en behandeling
Pulmonale hypertensie (PAH): herken kortademigheid, vermoeidheid en flauwvallen; lees over oorzaken, diagnose en effectieve behandelingen voor een betere levenskwaliteit.
Pulmonale hypertensie of PH is een aandoening waarbij er een hoge bloeddruk in de longen is. Deze aandoening maakt het moeilijk om te ademen. Sommige mensen met deze aandoening hebben extra zuurstof nodig. Door deze aandoening kan iemand ook duizelig worden en snel moe zijn. Sommige mensen met de aandoening vallen gemakkelijk flauw. De symptomen worden erger bij inspanning of hard werken. Pulmonale hypertensie is een ernstige aandoening, die dodelijk kan zijn. De aandoening maakt het moeilijker voor het hart om bloed te pompen. Omdat het hart harder moet werken, kan het ook ziek worden. Sommige mensen die erg ziek zijn, hebben een longtransplantatie of een hart-longtransplantatie nodig om te overleven. Pulmonale hypertensie heet voluit pulmonale arteriële hypertensie, hoewel de meeste mensen het pah, ph of pha noemen.
Afbeeldingengalerij
9 Afbeeldingen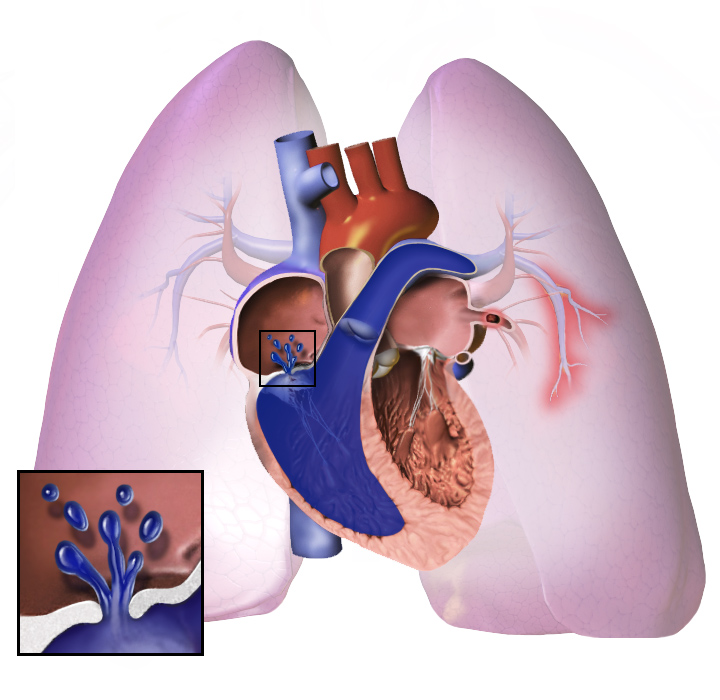






Wat is pulmonale hypertensie precies?
Pulmonale hypertensie is een verhoogde druk in de bloedvaten die het bloed van het hart naar de longen voeren. Dit maakt het werk voor het rechterdeel van het hart zwaarder. Belangrijk om te weten is dat pulmonale hypertensie (PH) een algemene term is: pulmonale arteriële hypertensie (PAH) is één type PH, maar er bestaan ook andere vormen met andere oorzaken. De klachten ontstaan geleidelijk en zijn in het begin vaak vaag.
Symptomen
- Kortademigheid bij inspanning, later ook in rust
- Snelle vermoeidheid en verminderd uithoudingsvermogen
- Pijn of druk op de borst
- Duizeligheid of flauwvallen (syncope), vooral bij inspanning
- Zwelling van enkels, voeten of buik door vochtopeenhoping
- Snelle of onregelmatige hartslag
De klachten kunnen langzaam erger worden. Omdat ze vaak op meer voorkomende ziekten lijken (zoals astma of hartfalen), kan de diagnose vertraging oplopen.
Oorzaken en risicofactoren
Pulmonale hypertensie kan veel verschillende oorzaken hebben. Enkele belangrijke oorzaken en risicofactoren zijn:
- Idiopathische of erfelijke oorzaken (geen duidelijke onderliggende ziekte)
- Aandoeningen waarmee PAH geassocieerd kan zijn, zoals bindweefselaandoeningen (bv. sclerodermie), hiv-infectie of leverziekten
- Chronische longziekten (bijv. COPD, longfibrose) die de longvaten beïnvloeden
- Chronische bloedstolsels in de longen (chronische trombo-embolische pulmonale hypertensie, CTEPH)
- Congenitale hartafwijkingen
- Bepaalde medicijnen en drugs (bijv. sommige anorectica of recreatieve drugs)
- Slaapapneu of andere aandoeningen die de zuurstofvoorziening beïnvloeden
Diagnose
Als uw arts PH vermoedt, volgt meestal een reeks onderzoeken om de oorzaak en de ernst te bepalen:
- Cardiale echografie (echocardiogram) om het hart en de druk te beoordelen
- Röntgenfoto of CT-scan van de borstkas
- Longfunctietesten en zuurstofmetingen
- Ventilatie-/perfusiescan (V/Q-scan) of CT-angiografie om chronische bloedstolsels op te sporen
- Bloedonderzoek (inclusief onderzoek naar onderliggende ziekten)
- In sommige gevallen slaaponderzoek (polysomnografie)
- Rechterhartkatheterisatie — dit is het gouden standaardonderzoek om de druk in de longslagaders precies te meten en de diagnose te bevestigen
Behandeling
De behandeling hangt af van de oorzaak en de ernst. Doelen van behandeling zijn klachtreductie, vertraging van ziekteprogressie en verbetering van kwaliteit van leven.
- Medicijnen gericht op de vaatspanning en bloedstroom in de longen: drie belangrijke klassen zijn endothelinereceptorantagonisten (bijv. bosentan, ambrisentan), fosfodiësterase-5-remmers (bijv. sildenafil, tadalafil) en prostacycline-analogen/agonisten (bijv. epoprostenol, treprostinil). Er zijn ook middelen zoals riociguat (sGC-stimulator) voor bepaalde vormen.
- Calciumkanaalblokkers — alleen bij patiënten die positief reageren op vasoreactiviteitstesten.
- Zuurstoftherapie bij chronische lage zuurstofwaarden.
- Diuretica om overmatig vocht te verminderen en zo de klachten te verlichten.
- Antistolling kan worden overwogen bij sommige vormen (bijvoorbeeld CTEPH), afhankelijk van oorzaak en risicoprofiel.
- Longoperaties: voor CTEPH kan een longendarteriëctomie (operatieve verwijdering van stolselmateriaal) genezend zijn. In ernstige, niet-behandelbare gevallen kan longtransplantatie of hart-longtransplantatie nodig zijn.
- Behandeling van onderliggende ziekten (bijvoorbeeld reumatische aandoeningen of HIV) is essentieel.
- Revalidatie en oefentherapie onder begeleiding verbetert vaak het uithoudingsvermogen en de kwaliteit van leven.
Leven met pulmonale hypertensie
- Volg de therapie en controleafspraken nauwkeurig op bij een gespecialiseerd centrum.
- Let op signalen van achteruitgang: toegenomen kortademigheid, meer zwelling, vaker flauwvallen.
- Vaccinaties (griep, pneumokokken) en stoppen met roken zijn belangrijk.
- Zwangerschap brengt grote risico's bij PAH en wordt meestal ontraden; bespreek dit met uw specialist bij kinderwens.
- Reizen en verblijf op grote hoogte kunnen extra risico’s geven; overleg met uw arts over voorzorgsmaatregelen en zuurstofbehoefte.
Prognose en follow‑up
De prognose varieert: vroegtijdige herkenning en behandeling verbeteren de uitkomst. Sommige mensen reageren goed op behandeling en blijven jarenlang stabiel; anderen hebben een progressieve ziekte ondanks therapie. Regelmatige controle in een gespecialiseerd centrum is belangrijk om behandeling aan te passen en complicaties vroeg te herkennen.
Wanneer contact opnemen met uw arts?
- Bij toenemende kortademigheid of vermoeidheid
- Bij herhaaldelijk flauwvallen of duizeligheid
- Bij snelle gewichtstoename door vochtophoping
- Bij vragen over medicatie, zwangerschap of plannen voor reizen
Als u meer wilt weten over uw specifieke situatie, is het raadzaam contact op te nemen met een longarts of een gespecialiseerd PH‑centrum. Zij kunnen onderzoeken doen, de oorzaak vaststellen en een behandelplan op maat aanbieden.
Tekenen en symptomen
Mensen met pulmonale hypertensie hebben moeite met ademhalen. Ze worden ook snel moe. Sommigen vallen ook gemakkelijk flauw. Ze kunnen pijn op de borst hebben. Sommige patiënten hebben zwelling van de voeten en enkels. Deze symptomen verergeren bij inspanning of zwaar werk.
Omdat veel ziekten de ademhaling kunnen bemoeilijken, moet een arts de achtergrond van de patiënt leren kennen. Dit helpt de arts om de patiënt te behandelen, zelfs als de patiënt een andere ziekte heeft. De arts doet ook verschillende tests. Pulmonale hypertensie laat het hart anders klinken. Een van de tests is het meten van de bloeddruk in de longslagader, het bloedvat dat van het hart naar de longen loopt.
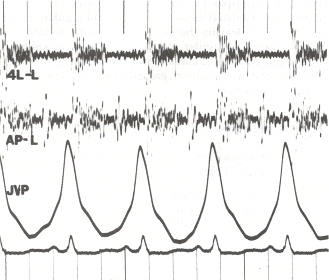
Om de oorzaak vast te stellen, zal de arts over het algemeen een grondige anamnese afnemen. Er wordt een gedetailleerde familieanamnese afgenomen om te bepalen of de ziekte misschien familiaal is. Een voorgeschiedenis van blootstelling aan cocaïne, methamfetamine, alcohol die leidt tot cirrose, en roken dat leidt tot emfyseem worden als belangrijk beschouwd. Bij lichamelijk onderzoek wordt gezocht naar typische tekenen van pulmonale hypertensie, zoals een luide P2 (pulmonaal klepsluitingsgeluid), (para)sternale heave, jugulaire veneuze zwelling, pedaal oedeem, ascites, hepatojugulaire reflux, knotten enz.
Wat gaat er mis met het lichaam
Bij pulmonale hypertensie worden de bloedvaten in de longen te nauw. De bloeddruk in de longen wordt hoog. Het hart werkt heel hard om het bloed door de nauwe bloedvaten te pompen. Later worden de bloedvaten in de longen hard en dik. Het hart moet harder werken.
Het hart kan zo hard werken dat het ziek wordt. Dit wordt hartfalen genoemd. Het zieke hart kan het bloed niet goed rondpompen. Er gaat minder bloed naar de longen, waardoor het bloed minder zuurstof krijgt. Hierdoor wordt het moeilijk om adem te halen. Dit wordt erger bij inspanning of hard werken.
Veroorzaakt
De meest voorkomende oorzaak van pulmonale hypertensie is linker hartfalen. Dit veroorzaakt pulmonale veneuze hypertensie. Dit leidt tot pulmonaal oedeem, of vochtophoping in de longen.
Veel ziekten kunnen pulmonale arteriële hypertensie (PAH) veroorzaken.
- Longziekten die ervoor zorgen dat het bloed minder zuurstof krijgt, zoals:
· chronisch obstructieve longziekte of COPD
· interstitiële longziekte
· Het Pickwickiaanse syndroom
- immuunsysteem problemen, zoals:
· AIDS
· sclerodermie
· andere auto-immuunziekten
- leverproblemen
· cirrose
· portale hypertensie
- andere oorzaken
· slaapapneu
· het slikken van pillen om af te vallen, zoals Fen-Phen, Aminorex, fenfluramine (Pondimin), en phentermine
· aangeboren hartaandoening
· schildklieraandoeningen,
· het nemen van drugs zoals cocaïne
· mogelijk Humaan herpesvirus 8
Wanneer een persoon pulmonale hypertensie heeft zonder andere oorzaak, wordt dit idiopathische pulmonale arteriële hypertensie of IPAH genoemd.
Wanneer er een familiegeschiedenis bestaat, wordt de ziekte familiaire pulmonale arteriële hypertensie, (FPAH) genoemd. IPAH en FPAH worden nu beschouwd als genetische aandoeningen die verband houden met mutaties in het BMPR2-gen, dat codeert voor een receptor voor botmorfogenetische proteïnen, en het 5-HT(2B)-gen, dat codeert voor een serotoninereceptor.
In de geneeskunde is pulmonale hypertensie (PH) een verhoging van de bloeddruk in de longslagader of het longvaatstelsel, die leidt tot kortademigheid, duizeligheid, flauwvallen en andere symptomen, die alle worden verergerd door inspanning. Afhankelijk van de oorzaak kan pulmonale hypertensie een ernstige ziekte zijn met een duidelijk verminderde inspanningstolerantie en rechtszijdig hartfalen. De ziekte werd voor het eerst vastgesteld door Dr. Ernst von Romberg in 1891. Er zijn vijf verschillende soorten: arteriële, veneuze, hypoxische, trombo-embolische of diverse.
Hoewel de termen primaire pulmonale hypertensie (dat wil zeggen door onbekende oorzaak) en secundaire pulmonale hypertensie (dat wil zeggen te wijten aan een andere medische aandoening) nog steeds voorkomen in materiaal dat onder patiënten en het grote publiek wordt verspreid, zijn deze termen in de medische literatuur grotendeels verlaten. Deze verandering is opgetreden omdat de oudere dichotome classificatie de pathofysiologie en het resultaat niet weerspiegelde. Zij leidde tot verkeerde therapeutische beslissingen, d.w.z. alleen "primaire" pulmonale hypertensie behandelen. Dit leidde op zijn beurt tot therapeutisch nihilisme voor veel patiënten met het etiket "secundaire" pulmonale hypertensie, en zou kunnen hebben bijgedragen tot hun overlijden. De term "primaire pulmonale hypertensie" is nu vervangen door "idiopathische pulmonale arteriële hypertensie". De termen "primaire" en "secundaire" pulmonale hypertensie mogen niet langer worden gebruikt. Nadere bijzonderheden zijn te vinden in het onderdeel Classificatie hieronder.
Veroorzaakt
De meest voorkomende oorzaak van pulmonale hypertensie is linkerhartfalen dat leidt tot pulmonale veneuze hypertensie. Dit kan te wijten zijn aan systolische of diastolische disfunctie van de linker hartkamer of aan valvulaire disfunctie zoals mitralisklepregurgitatie of mitralisstenose. Het manifesteert zich meestal als longoedeem.
Veel voorkomende oorzaken van pulmonale arteriële hypertensie (PAH) zijn HIV, sclerodermie en andere auto-immuunziekten, cirrose en portale hypertensie, sikkelcelziekte, aangeboren hartziekten, schildklieraandoeningen, en andere. Het gebruik van afslankpillen zoals Fen-Phen, Aminorex, fenfluramine (Pondimin), en phentermine heeft in het verleden geleid tot de ontwikkeling van PAH.
Pathogenese
Wat de oorspronkelijke oorzaak ook is, pulmonale hypertensie houdt in dat de bloedvaten die met en in de longen zijn verbonden, nauwer worden. Hierdoor wordt het moeilijker voor het hart om bloed door de longen te pompen, net zoals het moeilijker is om water door een smalle pijp te laten stromen dan door een brede pijp. Na verloop van tijd worden de aangetaste bloedvaten stijver en dikker, waardoor de bloeddruk in de longen verder toeneemt en de bloedstroom wordt belemmerd. Bovendien veroorzaakt de verhoogde belasting van het hart een verdikking en vergroting van de rechterhartkamer, waardoor het hart minder goed in staat is bloed door de longen te pompen, met rechterhartfalen tot gevolg. Omdat er minder bloed door de longen stroomt, krijgt de linkerkant van het hart minder bloed. Dit bloed kan ook minder zuurstof vervoeren dan normaal. Daarom wordt het steeds moeilijker voor de linkerkant van het hart om te pompen en de rest van het lichaam van voldoende zuurstof te voorzien, vooral tijdens lichamelijke activiteit.
Diagnose
Aangezien pulmonale hypertensie 5 grote typen kan hebben, moet een reeks tests worden uitgevoerd om pulmonale arteriële hypertensie te onderscheiden van veneuze, hypoxische, thombo-embolische of diverse varianten.
Bij lichamelijk onderzoek wordt gekeken naar typische tekenen van pulmonale hypertensie. Dit zijn onder andere veranderde hartgeluiden, zoals een wijd gespleten S2 of tweede hartgeluid, een luid P2 of pulmonaal klepsluitgeluid (onderdeel van het tweede hartgeluid), (para)sternale heave, mogelijk S3 of derde hartgeluid, en pulmonale regurgitatie. Andere tekenen zijn distensie van de vena jugularis (vergroting van de vena jugularis), perifeer oedeem (zwelling van de enkels en de voeten), ascites (zwelling van de buik door vochtophoping), reflux van de hepatojugularis, en knotten.
Verdere procedures zijn nodig om de aanwezigheid van pulmonale hypertensie te bevestigen en andere mogelijke diagnoses uit te sluiten. Deze omvatten doorgaans longfunctietests, bloedonderzoek, elektrocardiografie (ECG), arteriële bloedgasmetingen, röntgenfoto's van de borstkas (gevolgd door een CT-scan met hoge resolutie als interstitiële longziekte wordt vermoed), en ventilatie-perfusie of V/Q-scan om chronische trombo-embolische pulmonale hypertensie uit te sluiten. Een longbiopsie is gewoonlijk niet aangewezen, tenzij men denkt dat de pulmonale hypertensie te wijten is aan een onderliggende interstitiële longaandoening. Maar aan longbiopsies zijn risico's op bloedingen verbonden als gevolg van de hoge intrapulmonale bloeddruk. Klinische verbetering wordt vaak gemeten met een "zes-minuten-test", d.w.z. de afstand die een patiënt in zes minuten kan lopen. Stabiliteit en verbetering van deze meting correleren met een betere overleving.
Hoewel de pulmonale arteriële druk op basis van echocardiografie kan worden geschat, geeft drukbemonstering met een Swan-Ganz-katheter de meest definitieve meting. PAOP en PVR kunnen niet rechtstreeks met echocardiografie worden gemeten. Daarom is voor de diagnose van PAH een hartkatheterisatie nodig. Met een Swan-Ganz-katheter kan ook de cardiale output worden gemeten, die voor het meten van de ernst van de ziekte veel belangrijker is dan de pulmonale arteriële druk.
De normale pulmonale arteriële druk in een persoon die op zeeniveau leeft, heeft een gemiddelde waarde van 12-16 mm Hg (1600-2100 Pa). Er is duidelijk sprake van pulmonale hypertensie wanneer de gemiddelde druk in rust hoger is dan 25 mm Hg (3300 Pa). Als de gemiddelde druk in de longslagader bij inspanning tot boven 30 mm Hg (4000 Pa) stijgt, wordt dat ook als pulmonale hypertensie beschouwd.
De diagnose van PAH vereist de aanwezigheid van pulmonale hypertensie met twee andere voorwaarden. De occlusiedruk van de longslagader (PAOP of PCWP) moet lager zijn dan 15 mm Hg (2000 Pa) en de pulmonale vaatweerstand (PVR) moet groter zijn dan 3 Wood units (240 dyn-s-cm-5 of 2,4 mN-s-cm-5).
Classificatie
Huidige classificatie
In 2003 werd in Venetië het 3de Wereldsymposium over Pulmonale Arteriële Hypertensie bijeengeroepen om de classificatie te wijzigen op basis van het nieuwe inzicht in de ziektemechanismen. Het herziene systeem dat door deze groep werd ontwikkeld, vormt het huidige kader voor het begrijpen van pulmonale hypertensie.
Het systeem bevat verscheidene verbeteringen ten opzichte van het vroegere Evian-classificatiesysteem van 1998. De beschrijvingen van risicofactoren zijn geactualiseerd en de classificatie van congenitale systemische naar pulmonale shunts is herzien. Een nieuwe classificatie van genetische factoren in PH werd aanbevolen, maar niet uitgevoerd omdat de beschikbare gegevens ontoereikend werden geacht.
Het herziene classificatiesysteem van Venetië van 2003 kan als volgt worden samengevat:
- WHO groep I - Pulmonale arteriële hypertensie (PAH)
- WHO groep II - Pulmonale hypertensie geassocieerd met linker hartziekte
- WHO groep III - Pulmonale hypertensie geassocieerd met longziekten en/of hypoxemie
- WHO-groep IV - Pulmonale hypertensie ten gevolge van chronische trombotische en/of embolische aandoeningen
- WHO Groep V - Diversen
Vorige terminologie
De termen primaire en secundaire pulmonale hypertensie (PPH en SPH) werden vroeger gebruikt om de ziekte te classificeren. Dit leidde tot de veronderstelling dat alleen de primaire ziekte moet worden behandeld, en de secundaire variant moet worden genegeerd ten gunste van de behandeling van alleen de onderliggende ziekte. In feite zijn alle vormen van pulmonale arteriële hypertensie behandelbaar. Helaas is dit classificatiesysteem nog steeds aanwezig in de hoofden van veel artsen, en leidt het er waarschijnlijk toe dat veel patiënten geen behandeling krijgen. Deze nihilistische benadering van pulmonale arteriële hypertensie kan ook bijdragen tot onderdiagnose. Naar schatting zijn er in de VS ongeveer 100.000 patiënten met PAH, maar slechts 15-20.000 zijn gediagnosticeerd. Bij vele anderen is de verkeerde diagnose gesteld: COPD, astma of congestief hartfalen.
De term primaire pulmonale hypertensie (PPH) is nu in een groot deel van de medische literatuur vervangen door idiopathische pulmonale arteriële hypertensie (IPAH). Sommige artsen gebruiken echter nog steeds ten onrechte de oudere classificatie.
Epidemiologie
IPAH is een zeldzame ziekte met een incidentie van ongeveer 2-3 per miljoen per jaar en een prevalentie van ongeveer 15 per miljoen. Vrouwen hebben bijna driemaal zoveel kans om IPAH te krijgen als mannen.
Andere vormen van PAH komen veel vaker voor. Bij sclerodermie wordt de incidentie geschat op 6 tot 60% van alle patiënten, bij reumatoïde artritis tot 21%, bij systemische lupus erythematosus 4 tot 14%, bij portale hypertensie tussen 2 en 5%, bij HIV ongeveer 0,5%, en bij sikkelcelziekte variërend van 20 tot 40%.
Dieetpillen zoals Fen-Phen leverden een jaarlijkse incidentie op van 25-50 per miljoen per jaar.
Behandeling
De behandeling wordt bepaald door de vraag of de PH arterieel, veneus, hypoxisch, trombo-embolisch of divers is. Aangezien pulmonale veneuze hypertensie synoniem is met congestief hartfalen, bestaat de behandeling in het optimaliseren van de linker ventrikelfunctie door het gebruik van diuretica, bètablokkers, ACE-remmers, enz. of het herstellen/vervangen van de mitralisklep of de aortaklep.
Bij PAH worden veranderingen in de levensstijl, digoxine, diuretica, orale anticoagulantia en zuurstoftherapie als conventionele therapie beschouwd, maar van deze therapieën is nooit op een gerandomiseerde, prospectieve manier bewezen dat zij gunstig zijn.
Hoge doses calciumkanaalblokkers zijn nuttig bij slechts 5% van de IPAH-patiënten die vasoreactief zijn met een Swan-Ganz katheter. Helaas worden calciumkanaalblokkers in hoge mate verkeerd gebruikt, omdat ze worden voorgeschreven aan veel patiënten met nietvasoreactieve PAH, wat leidt tot een te hoge morbiditeit en mortaliteit.
Vasoactieve stoffen
Drie belangrijke routes zijn betrokken bij abnormale proliferatie en contractie van de gladde spiercellen van de longslagader bij patiënten met pulmonale arteriële hypertensie. Deze routes komen overeen met belangrijke therapeutische doelen bij deze aandoening en spelen een rol bij het bepalen welke van de drie klassen geneesmiddelen - endotheline receptor antagonisten, fosfodiësterase type 5 remmers en prostacyclinederivaten - zal worden gebruikt.
Prostacycline (prostaglandine I2) wordt algemeen beschouwd als de meest effectieve behandeling voor PAH. Epoprostenol (synthetische prostacycline, op de markt onder de naam Flolan®) wordt via een continu infuus toegediend, waarvoor een semi-permanente centrale veneuze katheter nodig is. Dit toedieningssysteem kan sepsis en trombose veroorzaken. Flolan® is onstabiel en moet daarom tijdens de toediening in ijs worden bewaard. Aangezien het een halfwaardetijd van 3 tot 5 minuten heeft, moet de infusie continu (24 uur per dag, 7 dagen per week) worden toegediend en kan onderbreking fataal zijn. Daarom zijn andere prostanoïden ontwikkeld. Treprostinil (Remodulin®) kan intraveneus of subcutaan worden toegediend, maar de subcutane vorm kan zeer pijnlijk zijn. Iloprost (Ilomedin®) wordt in Europa ook intraveneus gebruikt en heeft een langere halfwaardetijd. Iloprost (op de markt als Ventavis®) is de enige geïnhaleerde vorm van prostacycline die in de VS en Europa voor gebruik is goedgekeurd. Deze vorm van toediening heeft het voordeel van selectieve afzetting in de longen met minder systemische bijwerkingen.
De dubbele (ETA en ETB) endothelinereceptorantagonist bosentan (op de markt gebracht als Tracleer®) werd in 2001 goedgekeurd. Twee selectieve endothelinereceptorantagonisten (alleen ETA) bevinden zich in de laatste goedkeuringsfase: sitaxsentan en ambrisentan. Sildenafil, een selectieve remmer van cGMP-specifiek fosfodiësterase type 5 (PDE5), werd in 2005 goedgekeurd voor de behandeling van PAH. Het wordt voor PAH op de markt gebracht als Revatio®. Tadalafil (momenteel op de markt onder de naam Cialis® voor erectiestoornissen) bevindt zich momenteel in fase III van het onderzoek. Vasoactief intestinaal peptide door inhalatie zou in 2007 moeten beginnen met klinische proeven voor PAH. PRX-08066 is een serotonine-antagonist die momenteel wordt ontwikkeld voor hypoxische pulmonale hypertensie.
Chirurgisch
Atriumseptostomie is een chirurgische ingreep waarbij een verbinding tussen de rechter- en linkerhartboezem tot stand wordt gebracht. Het verlicht de druk op de rechterkant van het hart, maar ten koste van een lager zuurstofgehalte in het bloed (hypoxie). Deze procedure wordt het best uitgevoerd in ervaren centra. Longtransplantatie geneest pulmonale arteriële hypertensie, maar laat de patiënt achter met de complicaties van transplantatie, en een overleving van ongeveer 5 jaar.
Pulmonale tromboendarterectomie (PTE) is een chirurgische ingreep die wordt toegepast bij chronische trombo-embolische pulmonale hypertensie. Het is de chirurgische verwijdering van een georganiseerde trombus (stolsel) samen met de bekleding van de longslagader; het is een grote en zeer moeilijke ingreep die momenteel in een paar geselecteerde centra wordt uitgevoerd. Casereeksen tonen een opmerkelijk succes bij de meeste patiënten.
De behandeling van hypoxische en diverse vormen van pulmonale hypertensie is nog niet vastgesteld. In studies naar verschillende middelen worden momenteel echter patiënten opgenomen. Veel artsen zullen deze ziekten behandelen met dezelfde geneesmiddelen als voor PAH, totdat betere opties beschikbaar komen.
Prognose
Het NIH IPAH-register uit de jaren tachtig toonde een onbehandelde mediane overleving van 2-3 jaar na het tijdstip van diagnose, waarbij de doodsoorzaak meestal rechter ventrikelinsufficiëntie (cor pulmonale) was. Hoewel dit cijfer vaak wordt genoemd, is het vandaag de dag waarschijnlijk irrelevant. De resultaten zijn de laatste twee decennia drastisch veranderd. Dit kan het gevolg zijn van een nieuwere behandeling met geneesmiddelen, een betere algemene zorg en een vroegere diagnose (bias van de doorlooptijd). Uit een recent onderzoek naar de resultaten van patiënten die een behandeling met bosentan (Tracleer®) hadden gekregen, bleek dat 86% van de patiënten na 3 jaar nog in leven was. Nu er meerdere middelen beschikbaar zijn, wordt steeds vaker combinatietherapie toegepast. Het effect van deze middelen op de overleving is niet bekend, aangezien veel van deze middelen pas onlangs zijn ontwikkeld. Het zou niet onredelijk zijn te verwachten dat de mediane overleving in de nabije toekomst meer dan 10 jaar zal bedragen.
Vragen en antwoorden
V: Wat is pulmonale hypertensie of PH?
A: Pulmonale hypertensie of PH is een aandoening waarbij er een hoge bloeddruk in de longen is.
V: Wat zijn de symptomen van pulmonale hypertensie?
A: De symptomen van pulmonale hypertensie zijn onder andere ademhalingsmoeilijkheden, duizeligheid, vermoeidheid en flauwvallen.
V: Waarom hebben sommige mensen met pulmonale hypertensie extra zuurstof nodig?
A: Sommige mensen met pulmonale hypertensie hebben extra zuurstof nodig omdat ze door de aandoening moeilijk kunnen ademen.
V: Wanneer worden de symptomen van pulmonale hypertensie erger?
A: De symptomen van pulmonale hypertensie worden erger bij inspanning of hard werken.
V: Waarom is pulmonale hypertensie een ernstige aandoening?
A: Pulmonale hypertensie is een ernstige aandoening omdat het hart er meer moeite mee heeft om bloed rond te pompen en kan dodelijk zijn.
V: Wat is de volledige naam van pulmonale hypertensie?
A: De volledige naam van pulmonale hypertensie is pulmonale arteriële hypertensie, hoewel de meeste mensen het pah, ph, of pha noemen.
V: Wat hebben sommige erg zieke mensen met pulmonale hypertensie nodig om te leven?
A: Sommige zeer zieke mensen met pulmonale hypertensie kunnen een longtransplantatie of een hart-longtransplantatie nodig hebben om te blijven leven.
Gerelateerde artikelen
Auteur
AlegsaOnline.com Pulmonale hypertensie (PAH): symptomen, oorzaken en behandeling Leandro Alegsa
URL: https://nl.alegsaonline.com/art/80022
Bronnen
- ncbi.nlm.nih.gov : PMID 8692238
- ncbi.nlm.nih.gov : PMID 14985486
- ncbi.nlm.nih.gov : PMID 10555089
- ncbi.nlm.nih.gov : PMID 13679525
- ncbi.nlm.nih.gov : PMID 10903931
- ncbi.nlm.nih.gov : PMID 14659797
- ncbi.nlm.nih.gov : PMID 15194171