Creutzfeldt-Jakobziekte (CJD): prionen, symptomen en prognose
Leer over Creutzfeldt-Jakobziekte (CJD): oorzaken door prionen, herkenbare symptomen, ziekteverloop en prognose. Essentiële info voor patiënten en zorgverleners.
De ziekte van Creutzfeldt-Jakob (uitgesproken als KROITS-felt YAH-kohb) of CJD is een neurologische ziekte. Het is degeneratief (het wordt erger met de tijd); het kan niet worden genezen, en het veroorzaakt altijd de dood. CJD wordt soms een menselijke vorm van de "gekke-koeienziekte" (boviene spongiforme encefalopathie, of BSE) genoemd. BSE is in feite een oorzaak van één zeldzame vorm van de ziekte van Creutzfeldt-Jakob; de twee zijn niet dezelfde ziekte.
CJD wordt veroorzaakt door een infectieuze agens die prion wordt genoemd. Prionen zijn eiwitten die verkeerd gevouwen zijn. Prionen maken kopieën van zichzelf door correct gevouwen eiwitten te veranderen in verkeerd gevouwen vormen. CJD zorgt ervoor dat hersenweefsel zeer snel ongezond wordt. Terwijl de ziekte de hersenen vernietigt, ontstaan er gaten in de hersenen. De textuur van de hersenen verandert en wordt als een keukenspons.
Afbeeldingengalerij
6 Afbeeldingen




Vormen en oorzaak
Er bestaan meerdere vormen van CJD:
- Sporadische CJD — de meest voorkomende vorm; ontstaat zonder aanwijsbare oorzaak (ongeveer 85–90% van de gevallen).
- Familiaire (erfelijke) CJD — veroorzaakt door een mutatie in het PRNP-gen; familieleden kunnen verhoogd risico hebben.
- Iatrogene CJD — zelden overgedragen via besmet medisch materiaal (bijvoorbeeld onjuist gesteriliseerde neurochirurgische instrumenten, dura mater-transplantaten of vooraf gebruikte hormonen uit menselijke hypofyseweefsels).
- Variant CJD (vCJD) — geassocieerd met blootstelling aan BSE-gevaar (gevallen van vCJD zijn zeldzaam maar kregen veel aandacht tijdens BSE-uitbraken).
Symptomen
De ziekte begint doorgaans plotseling en verergert snel. Belangrijke klachten en verschijnselen zijn:
- Verlies van geheugen en cognitieve achteruitgang — verwarring, desoriëntatie, veranderingen in persoonlijkheid en moeite met denken.
- Bewegingsstoornissen — onhandigheid, stijfheid, spiertrillingen (myoclonieën), problemen met lopen en coördinatie (ataxie).
- Visuele problemen — wazig zien of verlies van gezichtsvermogen bij sommige patiënten.
- Psychiatrische symptomen — depressie, angst, prikkelbaarheid en soms hallucinaties.
- Slaapstoornissen — ernstige slaapverstoring komt vaak voor.
De combinatie en de snelheid van voortschrijding kunnen variëren per patiënt en per vorm van CJD.
Diagnose
De diagnose wordt gesteld op basis van klinische kenmerken en gespecialiseerde onderzoeken. Onderzoeksinstrumenten zijn onder andere:
- Neurologisch onderzoek — beoordeling van cognitieve functies, motoriek en reflexen.
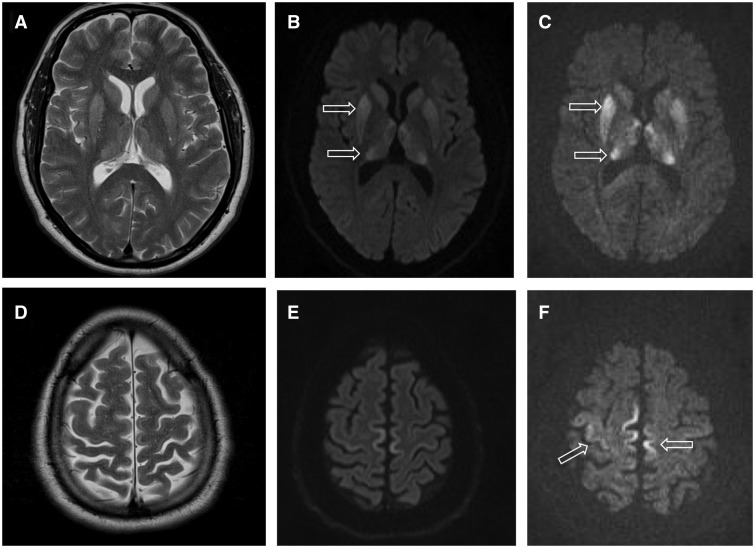
- MRI van de hersenen — kan karakteristieke afwijkingen laten zien, zoals hyperintensiteiten in de basale ganglia of thalamus.
- EEG (elektro-encefalogram) — kan periodieke scherpe golfcomplexen aantonen bij sommige vormen.
- Liquoronderzoek (CSF) — markers zoals 14-3-3-eiwit, verhoogde tau-eiwitwaarden of moderne tests zoals RT-QuIC kunnen helpen bij de bevestiging.
- Genetisch onderzoek — bij vermoeden van familiale CJD wordt het PRNP-gen getest.
Definitieve bevestiging van CJD kan alleen door neuropathologisch onderzoek (bijvoorbeeld hersenbiopsie of autopsie) worden geleverd, maar dit is vaak niet nodig of gewenst vanwege de risico's.
Behandeling en zorg
Er is momenteel geen genezende behandeling voor CJD. De zorg is primair ondersteunend en gericht op het verlichten van symptomen en het verbeteren van comfort. Mogelijke maatregelen:
- Pijnbestrijding en behandeling van spierspasmen of epileptische verschijnselen.
- Medicatie voor slaapstoornissen, angst of depressie waar nodig.
- Fysiotherapie, ergotherapie en logopedie om functioneren zo lang mogelijk te behouden en complicaties te beperken.
- Palliatieve zorg en psychosociale ondersteuning voor patiënt en familie, inclusief zorgplanning en rouwverwerking.
Onderzoek naar gerichte behandelingen is gaande, met benaderingen als remming van prionvorming, immunotherapie en genetische technieken, maar tot nu toe zijn er geen bewezen effectieve geneesmiddelen.
Besmettelijkheid en preventie
Prionen zijn bijzonder resistent tegen conventionele sterilisatiemethoden (zoals standaard autoclaveren). Daarom gelden in ziekenhuizen strikte voorzorgsmaatregelen bij operaties en procedures die hersen- of zenuwweefsel kunnen blootstellen. Preventieve maatregelen omvatten:
- Gebruik van wegwerpinstrumenten waar mogelijk of speciale sterilisatieprotocollen voor instrumenten die aan hoog-risicoweefsels zijn blootgesteld.
- Beperking van het gebruik van humane hypofysehormonen en zorg voor strenge bloed- en weefselveiligheidsprocedures.
- Surveillance en rapportage van gevallen door volksgezondheidsinstanties om mogelijke uitbraken of transmissieroutes te detecteren.
Prognose
De prognose bij CJD is slecht. De ziekte vordert meestal zeer snel; de meeste patiënten overlijden binnen enkele maanden tot een jaar na het begin van de symptomen. De mediane overleving voor sporadische CJD ligt typisch rond 4–6 maanden; minder dan 10% leeft langer dan twee jaar. Prognose kan variëren afhankelijk van de vorm van CJD en individuele factoren.
Frequentie en onderzoek
CJD is zeldzaam: de incidentie is ongeveer 1 geval per miljoen inwoners per jaar wereldwijd voor sporadische CJD. Familiaire vormen en vCJD zijn nog zeldzamer. Onderzoek naar de natuur van prionen, verbeterde diagnostische tests en mogelijke behandelingen gaat door in internationale onderzoeksnetwerken.
Ondersteuning voor patiënt en naasten
Gezien de snelle achteruitgang en de emotionele belasting is vroegtijdige betrokkenheid van een multidisciplinair team (huisarts, neuroloog, palliatief team, psycholoog, maatschappelijk werker) belangrijk. Families hebben vaak behoefte aan:
- Informatie over ziekteverloop en zorgopties.
- Praktische ondersteuning bij dagelijkse zorg en medische beslissingen.
- Emotionele en rouwbegeleiding.
Als u of een familielid symptomen heeft die lijken op die van CJD, neem dan contact op met uw huisarts of een neuroloog voor beoordeling en mogelijke doorverwijzing. Alleen een specialist kan verdere diagnostiek en advies over zorg en veiligheid geven.
Types en oorzaken van CJD
Types van CJD zijn:
- variant (vCJD):
Deze vorm van CJD kan worden veroorzaakt door het eten van voedsel met prionen erin, zoals vlees van koeien die BSE hebben ("gekkekoeienziekte"). Dit is echter een zeer zeldzame oorzaak van CJD.
- sporadisch (sCJD):
Dit is de meest voorkomende vorm van CJD. 85% van de gevallen van CJD is sporadische CJD. Niemand weet wat de oorzaak van sCJD is; het lijkt willekeurig te gebeuren.
- familiair (fCJD):
De meeste andere 15% van de gevallen van CJD zijn familiaire CJD. Dit is een vorm van CJD die in families voorkomt.
- iatrogeen:
Deze vorm van CJD wordt meestal veroorzaakt door een medische ingreep waarbij iemand bloed of weefsel krijgt van iemand met CJD. Iemand kan bijvoorbeeld iatrogene CJD krijgen als hij een bloedtransfusie of een hoornvliestransplantatie krijgt van iemand die CJD heeft.
Tekenen en symptomen
Het eerste symptoom van CJD is dementie, die zeer snel verergert. dementie veroorzaakt geheugenverlies, persoonlijkheidsveranderingen en hallucinaties.
Andere veel voorkomende mentale symptomen zijn:
- Anxiety
- Depressie
- Paranoia
- Obsessief-compulsieve symptomen
- Psychose
Lichamelijke symptomen van CJD zijn vaak:
- Problemen met spreken
- Schokkerige bewegingen (myoclonus)
- Problemen met het evenwicht (ataxie)
- Moeite met lopen
- Trillen of stijf zijn
- Problemen met zien
- Moeite met slikken, waardoor eten moeilijk of onmogelijk wordt
- Moeite met hoesten, wat longontsteking kan veroorzaken
- Bewegingen die de patiënt niet kan controleren (dyskinesie)
De meeste mensen met CJD sterven binnen zes maanden na het optreden van de eerste symptomen. Vaak sterven ze aan een longontsteking die wordt veroorzaakt door hoestproblemen. Ongeveer 15% van de patiënten overleeft twee jaar of langer. Sommige patiënten leven 4-5 jaar met voornamelijk geestelijke symptomen totdat de ziekte erger wordt en meer lichamelijke symptomen veroorzaakt. Zodra dit gebeurt, sterven mensen meestal binnen een jaar.
De symptomen van CJD worden veroorzaakt door het afsterven van steeds meer zenuwcellen in de hersenen. Wanneer wetenschappers hersenweefsel van een CJD-patiënt onder een microscoop bekijken, kunnen zij vele kleine gaatjes zien waar hele gebieden met zenuwcellen zijn afgestorven.
Diagnose
Artsen kunnen CJD vermoeden wanneer een persoon bepaalde symptomen heeft. Zo verergert dementie gewoonlijk langzaam. Dementie die zeer snel verergert is ongebruikelijk. Samen met symptomen zoals schokkerige bewegingen, kunnen deze symptomen wijzen op mogelijke CJD.
Er kunnen dan tests worden gedaan om aan te tonen of de persoon CJD heeft. Deze tests omvatten:
- Elektro-encefalografie (EEG): Deze test toont de elektrische activiteit in de hersenen. Een arts kan vaak veranderingen in het EEG zien die vaak voorkomen bij mensen met CJD. Het soort veranderingen dat op het EEG te zien is, hangt af van het type CJD dat de patiënt heeft en hoe ver de ziekte gevorderd is.
- Lumbaalpunctie (ruggenmergpunctie): Met deze test kan het hersenvocht (de vloeistof die de hersenen en het ruggenmerg omgeeft) worden onderzocht, op zoek naar een specifiek eiwit ("14-3-3-eiwit")
- MRI van de hersenen: Een test die gebruik maakt van een zeer sterke magneet om foto's van de hersenen te nemen
- Biopsie: Bij een biopsie neemt een chirurg met behulp van een naald een klein stukje weefsel uit het lichaam, zodat artsen het onder een microscoop kunnen bekijken. vCJD kan worden vastgesteld met een biopsie van de amandelen. Voor alle andere vormen van CJD is een biopsie van de hersenen de enige manier om met zekerheid te zeggen of iemand CJD heeft. Aangezien een biopsie van de hersenen echter hersenbeschadiging kan veroorzaken, wordt een biopsie van de hersenen meestal niet gedaan als uit andere tests al is gebleken dat iemand waarschijnlijk CJD heeft.

Behandeling
In 2016 is er nog geen behandeling die CJD geneest of zelfs de gevolgen ervan vertraagt. Er worden veel experimenten gedaan om te proberen behandelingen te vinden.
Vandaag de dag zijn de enige behandelingen voor CJD medicijnen die de symptomen van de ziekte behandelen en de patiënten helpen zich comfortabeler te voelen. Patiënten die epileptische aanvallen hebben, kunnen bijvoorbeeld anti-epileptica krijgen. Benzodiazepinen kunnen ervoor zorgen dat spierschokken minder vaak voorkomen.
Patiënten kunnen ook kiezen voor medische ingrepen om de symptomen te verlichten. CJD kan bijvoorbeeld zoveel problemen met slikken veroorzaken dat iemand niet meer kan eten. Sommige mensen met CJD kiezen ervoor om een voedingssonde te laten plaatsen als ze niet meer kunnen eten. Dit is een buisje dat in de maag gaat, zodat speciale vloeistof rechtstreeks in de maag kan worden gegeven om de persoon voeding te geven.
Verwante pagina's
- Prion
- Prionziekte
- Terminale ziekte
Vragen en antwoorden
V: Wat is de ziekte van Creutzfeldt-Jakob?
A: De ziekte van Creutzfeldt-Jakob (CJD) is een neurologische ziekte die degeneratief, ongeneeslijk en altijd fataal is.
V: Bestaat er een behandeling voor CJD?
A: Nee, er bestaat geen genezing voor CJD.
V: Waarom wordt CJD soms een menselijke vorm van "gekkekoeienziekte" genoemd?
A: CJD wordt soms een menselijke vorm van "gekkekoeienziekte" genoemd omdat boviene spongiforme encefalopathie (BSE), die een zeldzame vorm van CJD veroorzaakt, algemeen bekend staat als "gekkekoeienziekte".
V: Wat is de oorzaak van CJD?
A: CJD wordt veroorzaakt door een besmettelijk agens dat prion wordt genoemd. Dit is een verkeerd gevouwen eiwit dat kopieën van zichzelf kan maken door goed gevouwen eiwitten te veranderen in verkeerd gevouwen eiwitten.
V: Wat gebeurt er met het hersenweefsel bij CJD?
A: CJD zorgt ervoor dat het hersenweefsel zeer snel ongezond wordt, waardoor gaten in de hersenen ontstaan en de textuur van de hersenen verandert in die van een keukenspons.
V: Is BSE dezelfde ziekte als CJD?
A: Nee, BSE is niet dezelfde ziekte als CJD; het is eigenlijk een oorzaak van één zeldzaam type CJD.
V: Hoe veroorzaken prionen CJD?
A: Prionen veroorzaken CJD door zich verkeerd te vouwen en kopieën van zichzelf te maken ten koste van correct gevouwen eiwitten in de hersenen. Dit resulteert in de vernietiging van gezond hersenweefsel en de ontwikkeling van de gaten die kenmerkend zijn voor de ziekte.
Gerelateerde artikelen
Auteur
AlegsaOnline.com Creutzfeldt-Jakobziekte (CJD): prionen, symptomen en prognose Leandro Alegsa
URL: https://nl.alegsaonline.com/art/24152
Bronnen
- cdc.gov : "CJD (Creutzfeldt–Jakob Disease, Classic)"
- ncbi.nlm.nih.gov : "Creutzfeldt–Jakob disease: Transmissible spongiform encephalopathy; vCJD; CJD; Jacob-Creutzfeldt disease"
- bmj.com : "Bovine spongiform encephalopathy and variant Creutzfeldt–Jakob disease"
- doi.org : 10.1101/SQB.1996.061.01.052
- pubmed.ncbi.nlm.nih.gov : 9246478
- www3.interscience.wiley.com : "MM2-cortical-type sporadic Creutzfeldt–Jakob disease with early stage cerebral cortical pathology presenting with a rapidly progressive clinical course"
- doi.org : 10.1111/j.1440-1789.2008.00904.x
- pubmed.ncbi.nlm.nih.gov : 18410280
- who.int : who.int: "Fact sheets no 180: Variant Creutzfeldt-Jakob disease" Feb 2012 ed.
- prion.ucl.ac.uk : "Sporadic Prion Disease"
- doi.org : 10.1056/NEJM198610163151605
- pubmed.ncbi.nlm.nih.gov : 3762620
- prion.ucl.ac.uk : "Sporadic Prion Disease"
- merckmanuals.com : "Creutzfeldt–Jakob Disease (CJD)"
- ncbi.nlm.nih.gov : "Creutzfeldt-Jakob disease: Updated diagnostic criteria, treatment algorithm, and the utility of brain biopsy"